Interview
Anforderungen an Künstliche Intelligenz praktisch umgesetzt
KI und maschinelles Lernen revolutionieren die Medizinprodukte. Erfahren Sie, wie diese Technologien die Diagnostik verändern und welche regulatorischen Herausforderungen europäische Hersteller meistern müssen.

Regulatorische Anforderungen an KI-/ML-basierte Medizinprodukte
Die US-FDA veröffentlichte bereits Anfang 2021 einen „Artificial Intelligence and Machine Learning (AI/ML) Software as a Medical Device Action Plan„, die Benannten Stellen haben Ende 2023 die Version 5 ihres „Questionnaire „Artificial Intelligence (AI) in medical devices“ herausgegeben und in der EU wurde am 21. Mai 2024 der AI ACT verabschiedet und muss nun in nationales Recht umgesetzt werden.
Neben diesen Regularien gibt es bereits eine Reihe an Normen zu künstlicher Intelligenz, die aber zum größten Teil medizintechnisch-unspezifisch sind:
- ISO/TR 24291:2021 – Health informatics, Applications of machine learning technologies in imaging and other medical applications
- ISO/IEC 23894:2023 – Information technology – Artificial intelligence, Guidance on risk management
- ISO/IEC TR 24027:2021 – Information technology – Artificial intelligence (AI), Bias in AI systems and AI aided decision making
- ISO/IEC TR 24028:2020 – Information technology – Artificial intelligence, Overview of trustworthiness in artificial intelligence
- ISO/IEC TR 29119-11:2020 – Software and systems engineering – Software testing, Part 11: Guidelines on the testing of AI-based systems
- ISO/IEC TR 24029-1:2021 – Artificial Intelligence (AI) – Assessment of the robustness of neural networks, Part 1: Overview
- ISO/IEC 24029-2:2023 – Artificial intelligence (AI) – Assessment of the robustness of neural networks, Part 2: Methodology for the use of formal methods
- ISO/IEC 8183:2023 – Information technology – Artificial intelligence, Data life cycle framework
- IEEE 2801-2022 – IEEE Recommended Practice for the Quality Management of Datasets for Medical Artificial Intelligence
In naher Zukunft werden die Zulassungsverfahren sowie die regulatorischen Vorgaben für Prozesse und Medizinprodukte im EU- und US-Markt somit weitgehend konkretisiert sein. Eine frühzeitige Auseinandersetzung mit dem KI Act als europäischer Hersteller ist lohnenswert.
Wie sehen die notwendigen Anpassungen am QM-System und seinen Prozessen aus?
Um den für die Zulassung notwendigen Stand der Technik stets zu erfüllen, stellen sich Medizinprodukte-Hersteller einer besonderen Herausforderung. Denn die zugrunde liegenden Technologien entwickeln sich permanent weiter, während die regulatorischen Anforderungen oftmals hinterherhängen.
Folglich muss der Prozess zur Aktualisierung der regulatorischen Vorgaben deutlich an Dynamik gewinnen – von einem jährlichen auf mindestens einen quartalsweisen Zyklus. Dabei sind unter anderem folgende Datenquellen zur Ermittlung des jeweils aktuellen Stands der Technik zu berücksichtigen:
- Website der Europäischen Kommission zum Fortgang der europäischen AI-Gesetzgebung
- Website der FDA, Health Canada, der MHRA (UK) u.a. nationaler Behörden zu deren AI-Gesetzgebung (da diese die „Key Opinion Leader“ sind)
- Websites der Normenorganisationen ISO, IEC, IEEE zu Normen und Normentwürfen
- Website der Interessengemeinschaft der Notified Bodies (IG-NB) zu deren Positionierung
- Websites/Tools der Hersteller von KI-/ML-Technologie, bspw. Google AI
Im Qualitätsmanagementsystem müssen u.a. die folgenden Prozesse erweitert werden:
- Software-Lebenszyklusprozesse
- Risikomanagement
- Usability Engineering
- Datenmanagement
- Verifizierung & Validierung
- Klinische Bewertung
- Anwenderinformation/-kommunikation über das User Interface und die IFU
- Installations-, Trainings-, Wartungs- und Service-Prozesse
- Post Market Surveillance.
Wie steht es um die Erweiterung der Software-Lebenszyklusprozesse?
Das Datenmanagement spielt eine Schlüsselrolle für die Qualität und damit den Erfolg von Medizinprodukten mit KI- und Maschinellem Lernen. Dabei geht es um die Daten, die zum Training, zur Evaluierung/Validierung und für die spätere Optimierung des KI-Modells verwendet werden. Zu berücksichtigen ist hierbei, dass das EU-KI-Gesetz Medizinprodukte als Hochrisiko-KI-Systeme einstuft.
Konkret fordert Artikel 10 des Gesetzes:
- (2) Für Trainings-, Validierungs- und Testdatensätze gelten geeignete Daten-Governance- und Datenverwaltungsverfahren. […]
- (3) Die Trainings-, Validierungs- und Testdatensätze müssen relevant, repräsentativ, fehlerfrei und vollständig sein. Sie haben die geeigneten statistischen Merkmale, gegebenenfalls auch bezüglich der Personen oder Personengruppen, auf die das Hochrisiko-KI-System bestimmungsgemäß angewandt werden soll. […]
Zur Umsetzung dieser Anforderungen benötigen Medizintechnik-Unternehmen in der Regel zunächst Experten für Datenmanagement – eine Ressource, die in vielen Firmen bislang nicht zur Verfügung steht.
Darüber hinaus muss der Software-Entwicklungsprozess gemäß EN 62304 um spezifische Aktivitäten rund um das KI-Modell erweitert werden, wie die folgende Grafik zeigt:
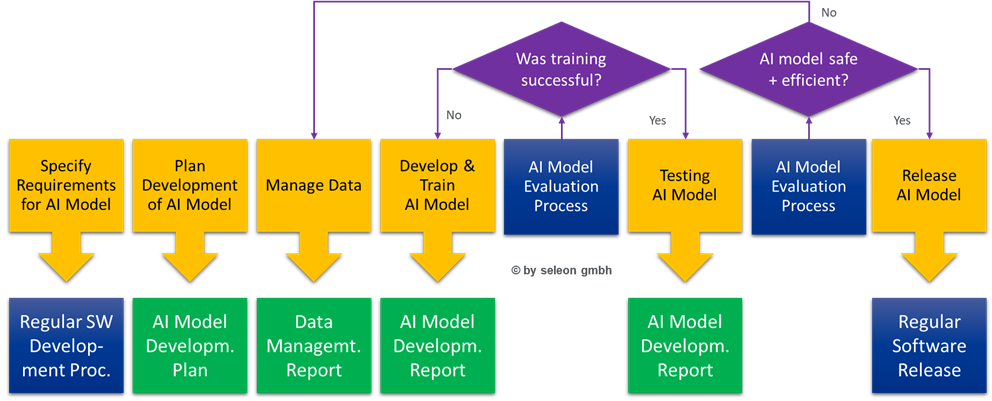
Abbildung 1 Zusätzliche KI-Modell-bezogene Aktivitäten im Software-Entwicklungsprozess
Nur so können Hersteller den regulatorischen Vorgaben für den Einsatz von KI und Maschinellem Lernen in Medizinprodukten gerecht werden.
Auch der Software-Wartungsprozess muss entsprechend erweitert werden, um die kontinuierliche Weiterentwicklung des KI-Modells regulatorisch korrekt unterstützen zu können.
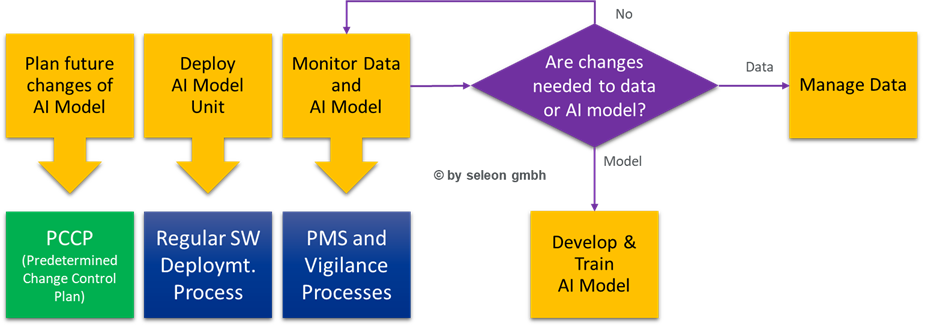
Abbildung 2 Zusätzliche KI-Modell-bezogene Aktivitäten im Software-Wartungsprozess
Ist auch eine Erweiterung des Usability Engineering erforderlich?
Die eher impliziten Anforderungen der Medizinprodukte-Verordnung (MDR) an die Gebrauchstauglichkeit (Usability) müssen insbesondere im Hinblick auf das für den Anwender oft intransparente Verhalten des KI-Modells im Medizinprodukt entsprechend angepasst werden.
Dies umfasst unter anderem die folgenden spezifischen Usability-Anforderungen für Medizinprodukte mit KI- und Maschinellem Lernen:
- Nutzungsumgebung-Gebrauchstauglichkeitstests: Es müssen Usability-Tests in einer Umgebung durchgeführt werden, die die tatsächliche klinische Arbeitsumgebung und die kognitive Belastung des Anwenders angemessen widerspiegelt.
- Workflow Management: Die Anforderungen an den klinischen Arbeitsablauf müssen identifiziert und in die KI-basierten Software-Spezifikationen für das medizinische Gerät umgesetzt werden.
- Nutzergruppe und Patientenpopulation: Die Anforderungen an die Benutzereingaben sowie die Zielpatientenpopulation müssen unter Berücksichtigung des Ausbildungsniveaus der Anwender spezifiziert werden.
- Transparenz: Die Systemausgaben müssen für den Anwender transparent sein und dessen Verständnisebene entsprechen.
- Erklärbarkeit: Die Datenverarbeitung und deren Bedeutung müssen für den Anwender erklärbar sein und dessen Ausbildungsstand berücksichtigen.
- Automatisierte Verzerrungen: Das Vorhandensein von automatisierten Verzerrungen (Automation Bias) muss untersucht werden.
- Fehlerbehandlung: Fehlermeldungen müssen für den Anwender transparent und verständlich sein. Außerdem muss eine Übergabe der Systemsteuerung an den Anwender möglich sein, wenn das System seinen Zweck nicht erfüllen kann.
- Update Management: Informationen zu Systemupdates (Modell oder Daten) müssen hinsichtlich Art, Grund und Auswirkungen auf Leistung und Sicherheit für den Anwender verständlich aufbereitet werden.
Praktische Hilfestellungen für die Umsetzung dieser Usability-Anforderungen finden sich auch in den „Good Machine Learning Practice for Medical Device Development: Guiding Principles“ der US-amerikanischen FDA, der kanadischen Health Canada sowie der britischen MHRA:
- Punkt 1 fordert multidisziplinäres Fachwissen während des gesamten Produktlebenszyklus:
Ein tiefgreifendes Verständnis der beabsichtigten Integration eines Modells in den klinischen Arbeitsablauf sowie des gewünschten Nutzens und der damit verbundenen Risiken für den Patienten kann dazu beitragen, dass ML-fähige medizinische Geräte sicher und wirksam sind und klinisch sinnvolle Anforderungen über den gesamten Lebenszyklus des Geräts erfüllen. - Punkt 7 legt den Schwerpunkt auf die Leistung des Mensch-KI-Teams:
Überlegungen zu menschlichen Faktoren und zur menschlichen Interpretierbarkeit der Modellergebnisse werden am besten berücksichtigt, wenn das Modell einen „Menschen in der (Rückkopplungs-)Schleife“ („Human in the loop“) hat, wobei der Fokus mehr auf der Leistung des Mensch-KI-Teams liegen sollte, als auf der Leistung des Modells für sich genommen. - Punkt 9 adressiert die Anwender-Informationen:
Den Anwendern werden klare, kontextbezogene Informationen zur Verfügung gestellt, die für die vorgesehene Zielgruppe (z. B. Leistungserbringer im Gesundheitswesen oder Patienten) geeignet sind. Dazu gehören der vorgesehene Verwendungszweck und die Indikationen für die Verwendung des Produkts, die Leistung des Modells für geeignete Untergruppen, die Merkmale der Daten, die zum Trainieren und Testen des Modells verwendet werden, akzeptable Eingaben, bekannte Einschränkungen, die Interpretation der Benutzeroberfläche und die Integration des Modells in den klinischen Arbeitsablauf. Die Anwender werden auch auf Geräteänderungen und Aktualisierungen aufgrund des Real-world Monitorings aufmerksam gemacht, auf die Grundlage für eine Entscheidungsfindung (Decision making) und die Möglichkeiten, dem Entwickler Bedenken zum Produkt mitteilen zu können
Die Erweiterung des Usability Engineering Prozesses um KI-Modell-bezogene Aktivitäten sähe wie folgt aus:
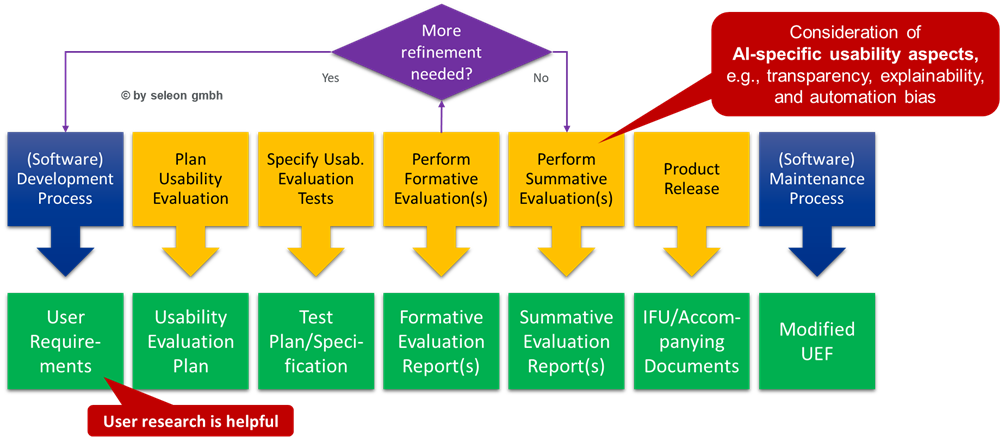
Abbildung 3 Zusätzliche KI-Modell-bezogene Aktivitäten im Usability Engineering
Wie erfolgt die Umsetzung in der klinischen Bewertung?
In der Klinischen Bewertung muss der Hersteller (jederzeit) nachweisen (können),
- dass das Medizinprodukt die vorgesehene Leistung erbringt,
- für seine Zweckbestimmung geeignet ist,
- sicher und wirksam ist,
- den klinischen Zustand oder die Sicherheit/Gesundheit von Patienten, Anwendern oder anderen Personen nicht gefährdet und
- ein positives Nutzen-Risiko-Verhältnis aufweist.
Die gilt natürlich auch für Medizinprodukte mit KI-/ML-Technologie, bei denen die folgenden spezifischen Anforderungen zu berücksichtigen sind:
- Im Plan zur klinischen Bewertung sind der Stand der Wissenschaft und Technik auf dem jeweiligen medizinischen Gebiet zu eruieren:
- Es muss der technische SOTA in Bezug auf AI identifiziert werden
- Der Nachweis des klinischen Bezugs dazu sowie der wissenschaftlichen Validität muss erbracht werden
- Der Plan für die Datenabfrage muss eine passende Datenbank-Recherche adressieren:
- Literatursuche in PubMed und ClinicalTrials.gov mit spezifischen medizinischen Fachbegriffen (MeSH), wie „Machine Learning“.
- Die Bewertung der klinischen Daten ist unter Berücksichtigung geeigneter Kriterien vorzunehmen:
- Analyse der Qualität von Literaturdaten zu klinischen Studien anhand der Kriterien von CONSORT-AI und SPIRIT-AI1
- Analyse der Qualität von Literaturdaten im Zusammenhang mit Studien zur diagnostischen Genauigkeit auf der Grundlage der Kriterien von STARD 2015
- Unabhängigkeit des Testdatensatzes vom Trainingsdatensatz (oder Information über Datensplitting) in Studien zur diagnostischen Genauigkeit
- Verwendung eines externen Testdatensatzes zusätzlich zu einem internen Testdatensatz in Studien zur diagnostischen Genauigkeit, um die Generalisierbarkeit des Modells zu testen
- Angemessenheit und nachvollziehbar korrekte Berechnung von Qualitätsmaßnahmen in klinischen Prüfungen und diagnostischen Genauigkeitsstudien
- Im Bericht zur klinischen Bewertung sind die Anforderungen an die Sicherheit und ein akzeptables Nutzen/Risiko-Profil sowie die Anforderungen an die Leistung zu belegen:
- Nachweis des angestrebten medizinischen Nutzens bei den festgelegten Werten der definierten Qualitätsmaßnahmen
- Vergleich des zu bewertenden Produkts mit klassischen klinischen Diagnose- oder Behandlungsverfahre (Referenzstandard)
- Nachweis der technischen/analytischen Leistungsfähigkeit
- Prospektive, randomisierte, multizentrische Studie nach dem Stand der Technik, um die Verallgemeinerbarkeit zu bestätigen und die Verwendung über den vorgesehenen Zweck hinaus zu untersuchen
- Nachweis der klinischen Leistungsfähigkeit in Bezug auf die Diskriminierungsleistung, Kalibrierungsleistung und die klinische Akzeptanz
Was ist also regulatorisch bei Medizinprodukten mit KI-/ML-Technologie zu berücksichtigen?
Wir fassen die wichtigsten Punkte aus diesem Beitrag nochmals wie folgt zusammen:
- Konformitätsbewertungsverfahren erfolgen in der EU gemäß der MDR, IVDR und später dem EU AI Act; in den USA erfolgen sie über die etablierten Zulassungsverfahren, mit technologie-spezifischen Besonderheiten
- Der Stand der Technik ändert sich ständig, was die Komplexität erhöht.
- Das Qualitätsmanagementsystem und seine Prozesse müssen für zusätzliche KI-/ML-Themen und -Anforderungen erweitert werden.
- Themen der Datenverarbeitung sind sehr wichtig, insbesondere die Unabhängigkeit der Testdaten von den Trainingsdaten. Qualifizierte Daten-Manager werden benötigt.
- Die Software-Lebenszyklusprozesse müssen um zusätzliche Aktivitäten erweitert und weitere Dokumente erstellt werden.
- Usability Engineering muss KI-/ML-spezifische Aspekte wie Transparenz, Erklärbarkeit und automatisierte Verzerrungen (Bias) berücksichtigen.
- Spezielle Anforderungen an Workflow Management, Patientenpopulation und Benutzergruppe müssen erfüllt werden.
- Die klinische Bewertung muss den KI-/ML-spezifischen Stand der Technik und die entsprechende Literatur berücksichtigen.
- Prospektive, randomisierte, multizentrische Studien nach dem neuesten Stand der Technik sind in der Regel Voraussetzung für eine erfolgreiche klinische Bewertung von Medizinprodukten mit KI/ML-Technologie, um Verallgemeinerbarkeit und Verwendung über den vorgesehenen Zweck hinaus zu bestätigen.
- Statische KI kann auf dem „klassischen Weg“ zugelassen werden, dynamische KI ist derzeit prinzipiell nicht zertifizierbar und statische Blackbox-KI kann durch eine Einzelfallentscheidung der Benannten Stelle zugelassen werden.
Sie wagen das Abenteuer KI/ML Technologie in Ihrem Medizinprodukt anzuwenden?
Wir begleiten Sie gerne dabei, Ihr Wissen und das Wissen Ihres Produktes aufzubauen und über den gesamten Lebenszyklus konform zu begleiten. Kontaktieren Sie uns oder besuchen Sie unsere Webseite für weiterführende Informationen!